
Teórica
1. De acordo com seus conhecimentos de Bioinformática e sobre alinhamento de sequências, responda se deve ser aplicado alinhamento global ou local nos exemplos abaixos
Muito usado para sequências com apenas alguns trechos conservados.
Resposta:
Alinhamento que retorna o melhor alinhamento ao longo de toda extensão.
Resposta:
Importante utilizá-lo quando duas sequências têm tamanhos próximos.
Resposta:
Alinhamento de alta similaridade em regiões do gene, as sequências adjacentes a estas regiões não entra na análise.
Resposta:
O BLAST é uma ferramenta no qual alinha trechos da sequência que produzam um escore elevado.
Resposta:
Importante utilizá-lo para alinhamento entre sequências de tamanhos diferentes.
Resposta:
2. MEGA é uma ferramenta integrada para realizar o alinhamento múltiplo automático e manual de sequências. Usando seus conhecimentos sobre o programa MEGA e observando o alinhamento feito abaixo, escolha a(s) alternativa(s) correta(s).
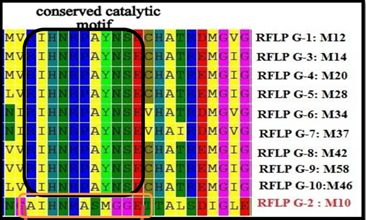
A) É possível alinhar sequências de DNA codificador por códons.
B) Alinhamentos múltiplos pelo MEGA podem ser feitas com o ClustalW, Muscle e Tcoffee
C) O MEGA possui integralidade com outras plataformas online como BLAST e GenBank
D) As sequências são conservadas apresentando um motivo conservados em 90% dos casos.
E) Não é possível afirmar com precisão qual evento mutagênico tenha acontecido com o fragmento RFLP G-2.
3. Avalie as afirmações sobre o quadro abaixo. O quadro apresenta sítios e a sequências de diferentes espécies do gênero Weedus.

https://www.cs.indiana.edu/~predrag/classes/2004falli400/lecture_notes_13.pdf
Existem duas alternativas corretas, marque as duas.
A) O sítio número 1 é o sitio mais informativo Segundo o método cladístico porque apresenta mais variação.
B) O sítio 3 não é um sítio informativo para reconstrução de filogenia porque não apresenta variação.
C) Os sítios 4 e 5 são sítios informativos Segundo o métodos cladísticos, entretanto eles apresentam informação diferente sobre a ancestralidade das espécies.
D) O sítio 6 é o sítio mais informativo para reconstrução fenética e cladística.
4. Com relação as árvores abaixo, responda

Tree equivalent
https://www.nature.com/scitable/topicpage/reading-a-phylogenetic-tree-the-meaning-of-41956
http://evolution.berkeley.edu/evolibrary/article/evo_07
http://epidemic.bio.ed.ac.uk/how_to_read_a_phylogeny
A) Todas as quatro árvores são não equivalentes
B) Todas as quatro árvores são equivalentes
C) Somente as árvores (i) e (iii) são equivalentes
D) Somente as árvores (iii) e (iv) são equivalentes.
E) Nenhuma resposta está correta.
5. No alinhamento abaixo estão representados as sequências de sete espécies. Quais das quatro afirmações abaixo você iria eliminar (afirmação incorreta) com base no conhecimento sobre alinhamento, evolução e reconstrução de filogenia.

https://www.mun.ca/biology/scarr/Transitions_vs_Transversions.html
A) Esse segmento alinhado é provavelmente parte de um pseudogene.
B) Esse trecho é mais provável de ser um segmento de RNA não codificaste.
C) Existem evidências que as transições ocorrem mais frequentes que as transversões
D) Existem evidências que as substituições sinônimas ocorrem mais frequentemente do que as não sinônimas.
6. As árvores abaixo foram propostas para as espécies do alinhamento. O método reconstrução filogenética de parcimônia (minimização do número de mutações) é usado para distinguir as quatro filogenias. Qual conclusão você iria descartar?
alinhamento questao arvore 6
árvores questão árvore 6

https://www.cs.indiana.edu/~predrag/classes/2004falli400/lecture_notes_13.pdf
A) A árvore 1 é a melhor arvore construída usando esse alinhamento segundo o método de parcimônia.
B) As árvores 1 e 2 são reconstruções melhores do que as árvores 3 e 4.
C) As árvores 1 e 3 são reconstruções melhores do que as árvores 2 e 4.
D) As árvores 1 e 4 são reconstruções melhores do que as árvores 2 e 3.
E) Nenhuma resposta está correta
7. O Heatmap abaixo foi construído usando genes diferencialmente expressos entre dois grupos de amostras após a análise de expressão por microarray. As linhas representam os diferentes genes, enquanto que as colunas representam as diferentes amostras. As cores azul e vermelho representam as amostras controle e amostra de interesse respectivamente.
Com relação a esse resultado.
Mapa de calor (Heatmap) para expressao de genes in microarray

http://www.opiniomics.org/you-probably-dont-understand-heatmaps/
A) Os genes usados para a construção do heatmap não apresentam diferença de expressao entre as amotras.
B) Podemos observar que as amostras não apresentam grande diferença de expressão através do padrão de corres da expressão de cada gene.
C) Podemos observar a separação dos dois grupos de amostras tanto pelo padrão de expressão dos genes quanto pela clusterização hierárquica representada acima do heatmap.
D) A separação das amostras com base na expressão desses genes permite a identificação de qual grupo a amostra é proveniente com confiança de 100%.
E) O conjunto de genes usado para a construção desse heatmap não foi adequadamente escolhido para evidenciar as diferenças entre as amostras.
F) Todas as respostas estão corretas.
8. Descreva a montagem de genoma referencia usando leituras (reads) provenientes de sequenciamento de nova geração.
9. Descreva a montagem de genoma de novo usando leituras (reads) provenientes de sequenciamento de nova geração.
10. Descreva o metodo de reconstrução filogenética baseado em distância (Neibor-joining).
11. Descreva o metodo de reconstrução filogenética baseado em caracteres (Parcimonia).
Prática
12) Realize uma busca por similaridade contra o banco de sequências nucleotídicas do NCBI usando o BLAST e retorne e descreva os resultados gráficos e resultados descritivos da sequência mais similar (primeira sequência subject) a sequência query. Descreva a sequencia query.
Sequência 1
13) Realize uma busca por similaridade contra o banco de sequências de proteína do NCBI usando o BLAST e retorne e descreva os resultados gráficos e resultados descritivos da sequência mais similar (primeira sequência subject) a sequência query. Descreva a sequencia query.
Sequência 1
14). Realizar a construção da rede de interação entre proteínas usando o banco de dados STRINGDB. Retornar a
rede de interação e explicar as análises estatísticas e de significancia da rede.
rede 1
rede 2
rede 3
rede 4
rede 5
rede 6
14.1) Reportar quais as 5 priemiras vias do KEGG (pathways) foram enriquecidas? Todas apresentam valor-p ajustado significativo considerando um alfa de 0.05?
14.2) Reportar quais os 5 priemiros GO (processos biológicos) foram enriquecidos? Todos apresentam valor-p ajustado significativo considerando um alfa de 0.05?