
Introdução
O programa Basic Local Alignment Search Tool (BLAST) foi desenvolvido para realizar buscas comparando sequências biológicas primárias contra um banco de dados que contém uma quantidade gigantesca de informação, retornando sequências mais similares e significância estatística da busca. A ferramenta BLAST é uma ferramenta muito utilizada dentro da bioinformática por ser uma ferramenta muito rápida e existirem diversas variações do BLAST, cada uma com um objetivo específico. Esse conjunto de variações de programas do pacote BLAST pode ser chamado de família BLAST.
Nesse tutorial vamos começar acessando a página do NCBI onde o BLAST está hospedado e vamos terminar usando o PUBMED, que é uma ferramenta para busca de artigos científicos. O tutorial tem como objetivo guiar buscas pelos diferentes programas BLAST e fornecer informações importantes que podem afetar o tipo de busca e seus parâmetros.
Ao final desse tutorial você será capaz de:
-
Submeter sequências biológicas primárias a busca por similaridade usando BLAST
-
Comparar as sequências usando as diferentes variações do BLAST
-
Interpretar os resultados
Os programas da família BLAST fazem busca por similaridade local entre sequências. Podemos usar tanto sequências de DNA quanto de proteína para realizar a busca.
BLAST – NCBI
Os programas BLAST estão hospedados no National Center for Biotechnology Information (NCBI) e tem ligação com o banco de dados desse centro. As buscas realizadas com o BLAST através da página do NCBI serão buscando sequências similares nesse banco de dados. Os programa BLAST realiza alinhamentos entre sequências de DNA ou proteínas chamadas query contra as sequências depositadas no banco de dados, essas sequências são chamadas subject.
Uma série de programas BLAST foram criados, permitindo a busca comparando sequências de proteína, ácidos nucleicos
Tabela 1. Diferentes variações do programa BLAST, tipo de query, tipo de subject e os diferentes propósitos. Adapted from “Bioinformatic for
Dummies”, Claverie and Notredame, 2007
Program
Query
Database
Usage
Choosing the BLAST
Blastn
DNA
DNA
Very similar DNA sequences (more than 70%)
I'm interested in non-coding and coding DNA
blastp
Protein
Protein
Similar protein sequences
I want to find similar proteins
blastx
TDNA
Protein
Analysis of the query DNA sequences
I want to discover proteins encoded in my query DNA sequence
tblastx
TDNA
TDNA
protein discovery and EST
I want to discovery new proteins
Os resultados da busca realizada pelos programas da família BLAST mostram sequências similares. Para essas sequências similares podemos questionar se o resultado é uma evidencia de sequências homologas (query e subject). A resposta é não. Entretatno podemos avaliar essa possibilidade. Para Avaliar essa possibilidade temos que observar a chance de uma alinhamento ser observado por acaso. Podemos fazer a comparaçao observando o alinhamento e comparar o resultado com:
-
Sequências reais mas não homólogas
-
Sequências reais que apresentam suas partes embaralhadas para presenvar propridades da composição
-
Sequências que são geradas aleatoriamente baseadas em modelos de DNA e proteínas
As análises estatíticas usam a terceira forma de análises, baseado nas comparações de sequências aleatórias e verificando quanto dessas comparações resultam em alinhamentos fortes.
Outras análises podem ser realizadas para verificar o quanto um alinhamento que não tem realação de similaridade apresentaria similaridade por acaso.
Estatística do alinhamento global
As comparações usando sequências aleatórias geradas com os modelos e sistemas de pontuação mais simples devem ser comparadas com uma distribuição de pontuação de alinhamentos ótimos. Entretanto essa distribuição não é conhecida. A comparação da significância estatística para os alinhamentos globais é realizada produzindo sequências aleatórias para calcular a pontuação do alinhamento ótimo.
Devido o comportamento da curva de distribuição de potuação do alinhamento global ser desconhecido não realizamos a comparação de média e desvio padrão dos valores de pontuação do alinhamento comparado com os resultados das sequências produzidas aleatoriamente, seria um erro transformar esses resultados assumindo uma aproximação da curva Z. Para avaliar a significância estatística usamos a quantidade de alinhamentos com sequências aleatórias produzem uma pontuação maior que a do alinhamento das sequências reais, se encontrarmos uma (1) comparação de 100 sequências aleatórias contendo pontuação maior que o alinhamento das sequências reais, dizemos que o valor p (p-value) é 0.01. Isso nos da uma ideia da significância do nosso alinhamento. Devemos ter cuidado quando fazemos múltiplas comparações (múltiplos testes) porque isso irá alterar o p-value, portanto alterando a significância dos resultados. Se selecionamos o melhor alinhamento com p-value de 0,0001 de uma conjunto de 1000 testes (comparações) independentes, teremos que corrigir o p-value para 0,1.
Estatística do alinhamento local
Os alinhamentos resultados do programa BLAST apresentam valores de pontuação e a esses valores é associado um valore chamado de expected-value (e-value). O e-value indica o número de alinhamentos que seriam esperados apresentando valores de score iguais ou melhores que o encontrado por acaso, dado o tamanho do banco de dados. O valore limite de corte do e-value, que determina o é significativo, é inserido pelo usuário e vai filtrar quais alinhamentos serão apresentados como resultado da busca. O e-value padrão é igual a 10, quanto maior o valor limite do e-value, menor será a restrição. Valores altos para o e-value fazem com que nenhuma informação biológica seja perdida comparado quando usamos valores iniciais muito restritos. Frequentemente valores limites do e-value são trocados por 10-6 ou menores para fazer uma busca bem restrita.
Ao contrário da estatística dos valores dos alinhamentos globais, a estatística dos valores dos alinhamentos locais é muito melhor compreendida. Vamos considerar primeiro os alinhamentos sem gaps, que correspondem a primeira versão do BLAST.
Um alinhamento sem gaps é simplesmente uma comparação de duas sequências do mesmo tamanho. Nessa comparação iremos procurar pelas maiores pontuaçoes entre os pares de segmentos (high-scoring segment pairs – HSPs) que são segmentos onde a pontuaçao do alihamento não aumenta comparando um trecho maior das sequências. A análise dos HSPs é feita comparando o e-value obtido em um HSP com os obtidos em sequências formadas aleatoriamente. A sequência aleatória pode ser gerada escolhendo aminoácidos aleatórios de forma independente com base em uma frequências de aminoácidos.
Comparações de sequências longas de aminoácidos apresentam pontuaçoes altas, mesmo sendo sequências produzidas de forma aleatória. Então não podemos usar somente os valores de pontuação, esperando que por causa das comparações dos aminoácidos sejam negativas também teriamos comparações aleatórias negativas.
O número de HSPs esperados com um valor e pontuação de pelo menos S é chamado de e-value. O número de HSPs comparando duas sequências depende do tamanho das duas sequências (m x n), se dobrarmos o tamanho de m estaremos dobrando o número de HSPs possíveis.
A redução do valor e-value é exponencial com o aumento do valor score para uma HSP de tamanho m x n (Figura 1).
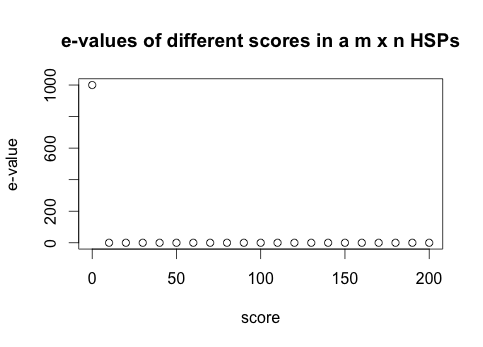
Figura 1. Valor do e-value (eixo y) para uma comparação de duas sequências tamanho m x n com diferentes scores (eixo x) . O e-value diminui rapidamente quando o score aumenta.
Essas comparações estão considerando somente a sequência m x n. Para considerarmos a comparação de todo o banco e dados devemos fazer alguma considerações. Primeiro consideramos que todas as proteínas tem o mesmo peso (priori) para serem relacionadas com a sequência m. O valor e-value obtido para a comparação de uma sequência m contra um pequeno banco de dados é diferente do mesmo valor e-value quando comparamos a sequência m com um grande banco de dados. Essa correção pode ser feita multiplicando o valor do e-value contra o número de sequências contidas nos dois bancos, assim teremos o quanto significativo esses valore e-value são.
Outra forma de fazer essa correção é colocar pesos diferentes para as sequências, onde sequencias maiores terão uma chance maior do que as seqências menores de conterem sequências realcionadas com a sequencia m (query). Considerando também o banco de dados como uma única sequência podemos fazer a realação do e-value e tamanho do banco de dados.
Formatos das sequências query e banco de dados
As sequências usadas pelo BLAST são sequências em um formato muito simples. As informações sobre as sequências, tanto DNA quanto proteínas, são aramazenadas no formado de apenas uma letra para cada nucleitideos (A, T, C e G) ou aminoácidos. As informações sobre cada sequência query e subject no banco de dados são armazenadas no formato FASTA, onde cada sequência começa com o sinal “>” seguindo do identificador único, e de outras informações adicionais sobre a sequência (essas informações adicionais são opcionais). A próxima linha contém a sequência (DNA ou proteína). O banco de dados usado pelo BLAST é um banco binário indexdo com os arquivos FASTA, o banco é criado pelo programa formatdb e depois podemos usar o programa BLAST.
Figura 2. Sequência no formato FASTA.
Alinhamentos
Os alinhamentos são pareamentos de duas sequências onde uma letra em uma sequência (corresposndente de um nucleotídeo ou aminoácido) é pareado com outra letra ou gap (-) da outra sequência. Cada alinhamento possui uma valor de pontuação (score) que indica o quanto o alinhamento tem posições similares/idênticas. Esse score é calculado por algoritmos que computam o valor do pareamento de cada letra no alinhamento e depois somam esses valores. O cálculo do score de cada alinhamento é feito com base no tipo de sequência, proteína ou DNA.
Matrizes de susbtituição
Dayhoff produziu um conjunto de matrizes de substuição para serem usadas na comparação de sequências de proteína. Dayhoff construiu a matriz PAM (“point accepted mutation”) para modelagem de evolução molecular através da comparação de proteínas relacionadas e contabilizando a porcentagem de mudanças que aconteceram. PAM 1 correspoende a mudança de 1% de todos os aminoácidos na proteína. Depois de 100 PAMs de evolução nem todos os aminoácidos terão mudado, alguns não irão mudar, enquanto outros irão mudar diversas vezes, inclusive voltando para o estado original. Por isso podemos reconhecer proteínas homólogas com mais de 100 PAMs de evolução. Não existe correspondência do tempo evolutivo e as matrizes PAM, já que proteínas evoluem em taxas diferentes. Quando duas sequências são comparadas não sabemos a priori qual matriz irá explicar a similaridade que elas podem ter. A matriz mais usada para fazer a comparação entre duas sequencias de aminoácidos é a matriz PAM-250. Depois outras matrizes PAM foram publicadas mas elas não diferem muito da matriz PAM250.
Henikoff & Henikoff usaram comparações de alinhamentos de sequências relacionadas, mas que apresentam distantes, ao contrário das comparacões que levaram a produção da matriz PAM que usou sequências realacionadas próximas. Essa abordagem criou o conjunto de matrizes BLOSUM que são consideradas serem superiores para encontrarem relaçoes biológicas entre proteínas. Entretanto esse conjunto de matrizes não produz um modelo evolutivo. O programa BLAST utiliza para comparações de sequências de proteína a matrix BLOSUM62 como padrão, entretanto outras matrizes da família BLOSUM ou PAM podem ser usadas.
Figura 3. Matriz BLOSUM62. http://upload.wikimedia.org/wikipedia/commons/5/52/BLOSUM62.gif
Para comparações de nucleotídeos usamos os valores +1 para as combinações de letras no alinhamento (match) e -2 para não combinações (mismatch).
Os gaps também tem um custo negativo (penalidade) para abertura (gap-creation ou gap-opening) e outro custo para extensão de um gap existente (gap-extension). Os valores de criação (penalidade) do gap são maiores que os valores de extensão do gap.
Algoritmo BLAST
O programa BLAST realiza a busca usando de seguimentos da sequência query contra as sequências do banco de dados. A sequência query é dividida em pequenas palavras (word) que serão usadas na busca. Essas subsequências são comparadas com as sequências subjects até encontrar sequências idênticas. Após encontrarem uma região idêntica a subsequência é extendida nas duas direções da subsequência com outras partes da sequência query, verificando se a similaridade entre as duas sequências continua para produzir um alinhamento entre as duas sequências. A extensão do alinhamento continua enquanto o valore de pontuaçao continuar aumentando (presença de matches), até que o alinhamento não apresente mais similaridade e a pontuação caia abaixo de um valor limite (dropoff) devido a presença de vários mismatches. O tamanho da subsequência (wordsize) é configurado pelo usuário, sendo os valores padrões são 28 para DNA e 3 para proteínas.
Referencias
Dayhoff, M. O.; Schwartz, R. M.; Orcutt, B. C. (1978). “A model of evolutionary change in proteins”. Atlas of Protein Sequence and Structure 5 (3): 345–352.
Henikoff, S.; Henikoff, J.G. (1992). “Amino Acid Substitution Matrices from Protein Blocks”. PNAS 89 (22): 10915–10919. doi:10.1073/pnas.89.22.10915. PMC 50453. PMID 1438297