Testando a confiança dos ramos da árvores
Vamos usar aqui nesse tutorial dois testes de para avaliar a confiança dos ramos encontrados em uma árvore filogenética. Usaremos os teste de bootstrap, o mais usado para avaliar a confiança de uma árvore, e o teste interior-branch.
Teste bootstrap
Carrege o arquivo “Chloroplast_Martin.meg” e depois selecione na barra de ferramentas “Phylogeny –> Construct/Test Maximum Parsimony Tree ”. A janela “Analysis Preferences” irá aparecer para nós selecionarmos “Model/Method” e o teste de confiança da árvore (“Test of Phylogeny”). Vamos selecionar o “Model/Method” k2p e o teste de bootstrap.
Clicar em “Compute” e logo aparecerá uma janela com indicativo de progresso da análise em forma de barra. Se você estiver trabalhando com muitas sequências ou sequências muito longa pode ir tomar uma café e voltar mais tarde. Alguns teste boostrp podem demorar muito dependendo da quantidade de dados você tem.
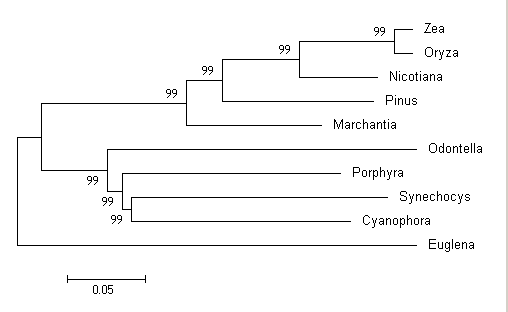
Após a análise terminar irá surgir a janela “Tree Explorer” onde vai aparecer o resultado da sua árvore com os valores de bootstrap para cada nó. Nó representa o ponto de divergência entre as sequências usadas. Na janela “Tree explorer” teremos duas abas chamadas “original tree” e “Bootstrap consensus tree”.
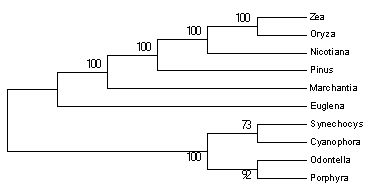
Figura 1.
Podemos produzir uma árvore condensada (condensed tree) através do menu principal do “Tree Explorer” –> “Compute” –> “Condensed Tree”.
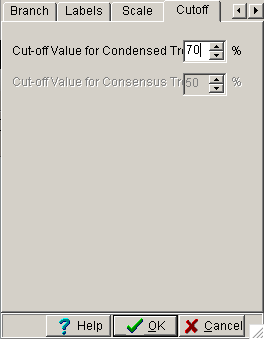
Figura 02.
Para mudar o ponto de corte do valor bootstrap devemos acessar no menu pricipal “View” –> “Options” e selecione na aba cutoff o valor de 80%. Irá aparecer uma árvore onde somente o valor de 80 ou acima são suportados. Caso deseje ver novamente a árvore original podemos ir no menu principal “Compute”–> “Condensed Tree”.
Figura 3.
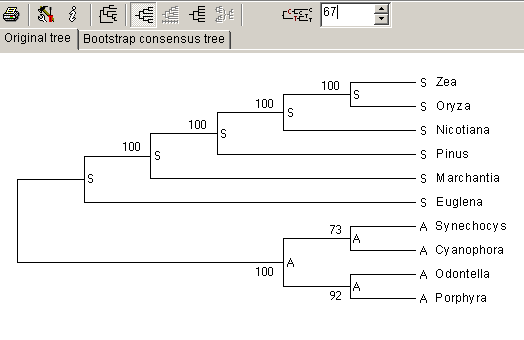
Figura 4. Representação do estado do caracter 67 em cada um dos nós.
Para salvar essa árvore devemos selecionar no menu principal “Image”– > “ Save as PDF File”. Podemos ainda salvar a árvore com os formatos EMF ou PNG.
Teste Interior-branch
Para as árvores do tipo NJ podemos usar o testeInterior-branch para computar o erro padrão para cada ramo interno da árvore. Novamente carregue o arquivo “Chloroplast_Martin.meg” se ele já não estiver carregado, selecione na barra de ferramentas Phylogeny –> Construct/Test Neighbor-Joining Tree ”. A janela “Analysis Preferences” irá aparecer para nós selecionarmos “Model/Method” e o teste de confiança da árvore (“Test of Phylogeny”). Vamos selecionar o “Model/Method” “p-distance” e o teste de Interior-branch.
Podemos fazer os ramos que não tem suporte ficarem sem no apresentando dicotomia, mostrando que a relação daquele ramo não está definida e não está claro qual sequências seria a mais relacionada. Não temos confiança para afirmar qual seria a mais próxima.
Após os cálculos do teste a árvore será exibida da mesma forma que no teste anterior. Como usamos um métodos de distância para produzir a nossa árvore não poderemos mostrar para cada sítio o estado de cada nó ou ramo.